Search Results for author:
Found 11 papers, 11 papers with code
Accelerating Inference in Molecular Diffusion Models with Latent Representations of Protein Structure
Diffusion generative models have emerged as a powerful framework for addressing problems in structural biology and structure-based drug design.
PLANTAIN: Diffusion-inspired Pose Score Minimization for Fast and Accurate Molecular Docking
Traditional docking methods predict ligand poses by minimizing a physics-inspired scoring function.

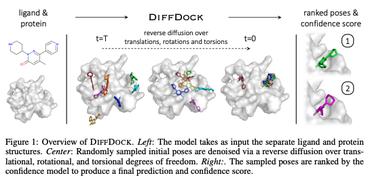
Generating 3D Molecules Conditional on Receptor Binding Sites with Deep Generative Models
The goal of structure-based drug discovery is to find small molecules that bind to a given target protein.

MOLUCINATE: A Generative Model for Molecules in 3D Space
Recent advances in machine learning have enabled generative models for both optimization and de novo generation of drug candidates with desired properties.
Learning a Continuous Representation of 3D Molecular Structures with Deep Generative Models
Machine learning in drug discovery has been focused on virtual screening of molecular libraries using discriminative models.
Generating 3D Molecular Structures Conditional on a Receptor Binding Site with Deep Generative Models
We show that valid and unique molecules can be readily sampled from the variational latent space defined by a reference `seed' structure and generated structures have reasonable interactions with the binding site.
SidechainNet: An All-Atom Protein Structure Dataset for Machine Learning
Despite recent advancements in deep learning methods for protein structure prediction and representation, little focus has been directed at the simultaneous inclusion and prediction of protein backbone and sidechain structure information.
libmolgrid: GPU Accelerated Molecular Gridding for Deep Learning Applications
There are many ways to represent a molecule as input to a machine learning model and each is associated with loss and retention of certain kinds of information.

Visualizing Convolutional Neural Network Protein-Ligand Scoring
Here we present three methods for visualizing how individual protein-ligand complexes are interpreted by 3D convolutional neural networks.
Ligand Pose Optimization with Atomic Grid-Based Convolutional Neural Networks
Docking is an important tool in computational drug discovery that aims to predict the binding pose of a ligand to a target protein through a combination of pose scoring and optimization.
Protein-Ligand Scoring with Convolutional Neural Networks
A CNN scoring function automatically learns the key features of protein-ligand interactions that correlate with binding.
