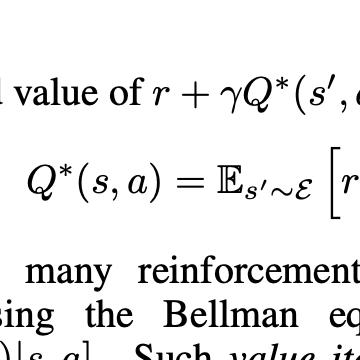Benchmarking Deep Graph Generative Models for Optimizing New Drug Molecules for COVID-19
Design of new drug compounds with target properties is a key area of research in generative modeling. We present a small drug molecule design pipeline based on graph-generative models and a comparison study of two state-of-the-art graph generative models for designing COVID-19 targeted drug candidates: 1) a variational autoencoder-based approach (VAE) that uses prior knowledge of molecules that have been shown to be effective for earlier coronavirus treatments and 2) a deep Q-learning method (DQN) that generates optimized molecules without any proximity constraints. We evaluate the novelty of the automated molecule generation approaches by validating the candidate molecules with drug-protein binding affinity models. The VAE method produced two novel molecules with similar structures to the antiretroviral protease inhibitor Indinavir that show potential binding affinity for the SARS-CoV-2 protein target 3-chymotrypsin-like protease (3CL-protease).
PDF Abstract