DeepGS: Deep Representation Learning of Graphs and Sequences for Drug-Target Binding Affinity Prediction
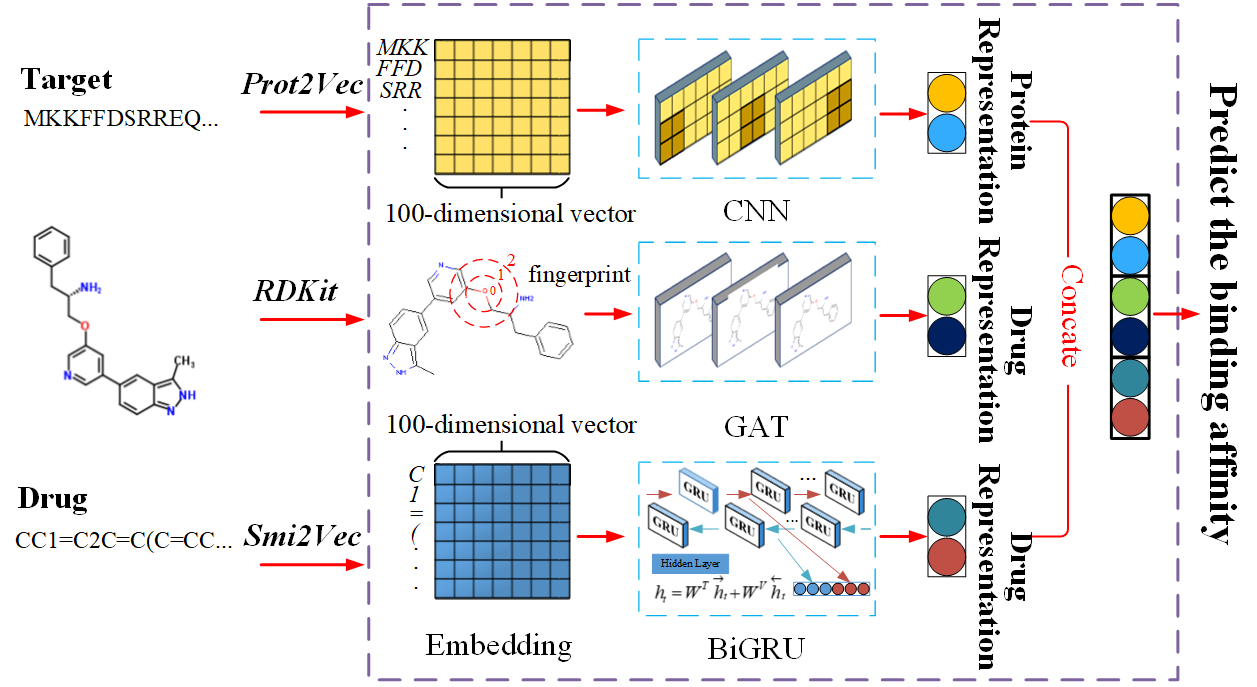
Accurately predicting drug-target binding affinity (DTA) in silico is a key task in drug discovery. Most of the conventional DTA prediction methods are simulation-based, which rely heavily on domain knowledge or the assumption of having the 3D structure of the targets, which are often difficult to obtain. Meanwhile, traditional machine learning-based methods apply various features and descriptors, and simply depend on the similarities between drug-target pairs. Recently, with the increasing amount of affinity data available and the success of deep representation learning models on various domains, deep learning techniques have been applied to DTA prediction. However, these methods consider either label/one-hot encodings or the topological structure of molecules, without considering the local chemical context of amino acids and SMILES sequences. Motivated by this, we propose a novel end-to-end learning framework, called DeepGS, which uses deep neural networks to extract the local chemical context from amino acids and SMILES sequences, as well as the molecular structure from the drugs. To assist the operations on the symbolic data, we propose to use advanced embedding techniques (i.e., Smi2Vec and Prot2Vec) to encode the amino acids and SMILES sequences to a distributed representation. Meanwhile, we suggest a new molecular structure modeling approach that works well under our framework. We have conducted extensive experiments to compare our proposed method with state-of-the-art models including KronRLS, SimBoost, DeepDTA and DeepCPI. Extensive experimental results demonstrate the superiorities and competitiveness of DeepGS.
PDF Abstract


 2D-3D-S
2D-3D-S
