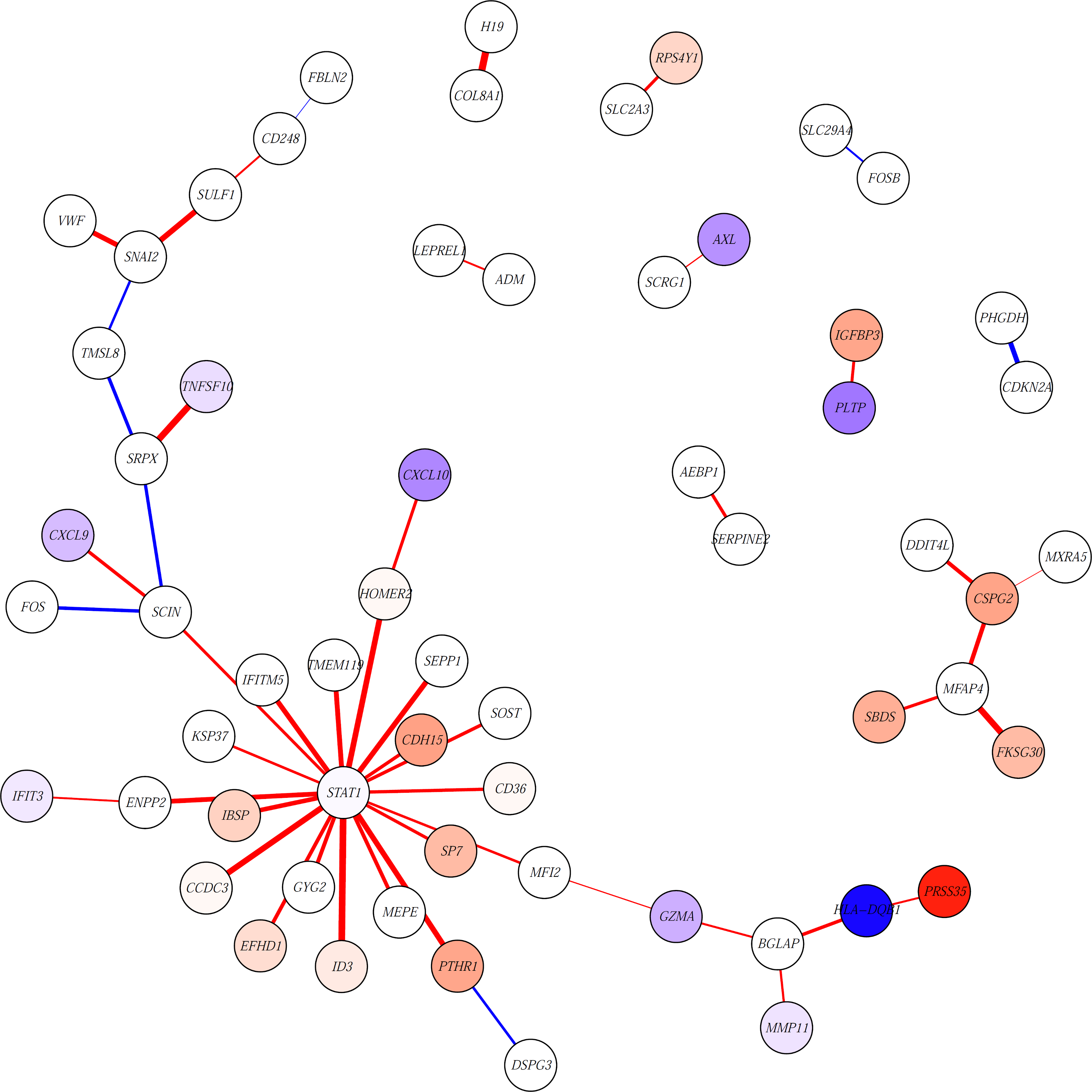
Estimating sample-specific regulatory networks
Biological systems are driven by intricate interactions among the complex array of molecules that comprise the cell. Many methods have been developed to reconstruct network models of those interactions. These methods often draw on large numbers of samples with measured gene expression profiles to infer connections between genes (or gene products). The result is an aggregate network model representing a single estimate for the likelihood of each interaction, or "edge," in the network. While informative, aggregate models fail to capture the heterogeneity that is represented in any population. Here we propose a method to reverse engineer sample-specific networks from aggregate network models. We demonstrate the accuracy and applicability of our approach in several data sets, including simulated data, microarray expression data from synchronized yeast cells, and RNA-seq data collected from human lymphoblastoid cell lines. We show that these sample-specific networks can be used to study changes in network topology across time and to characterize shifts in gene regulation that may not be apparent in expression data. We believe the ability to generate sample-specific networks will greatly facilitate the application of network methods to the increasingly large, complex, and heterogeneous multi-omic data sets that are currently being generated, and ultimately support the emerging field of precision network medicine.
PDF Abstract