Graph Dynamical Networks for Unsupervised Learning of Atomic Scale Dynamics in Materials
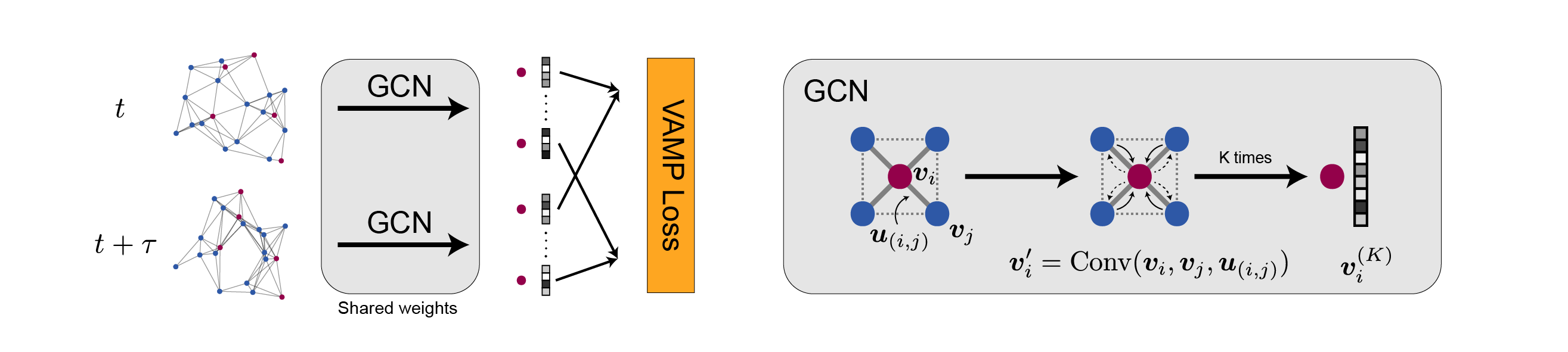
Understanding the dynamical processes that govern the performance of functional materials is essential for the design of next generation materials to tackle global energy and environmental challenges. Many of these processes involve the dynamics of individual atoms or small molecules in condensed phases, e.g. lithium ions in electrolytes, water molecules in membranes, molten atoms at interfaces, etc., which are difficult to understand due to the complexity of local environments. In this work, we develop graph dynamical networks, an unsupervised learning approach for understanding atomic scale dynamics in arbitrary phases and environments from molecular dynamics simulations. We show that important dynamical information can be learned for various multi-component amorphous material systems, which is difficult to obtain otherwise. With the large amounts of molecular dynamics data generated everyday in nearly every aspect of materials design, this approach provides a broadly useful, automated tool to understand atomic scale dynamics in material systems.
PDF Abstract
