Learning from the Density to Correct Total Energy and Forces in First Principle Simulations
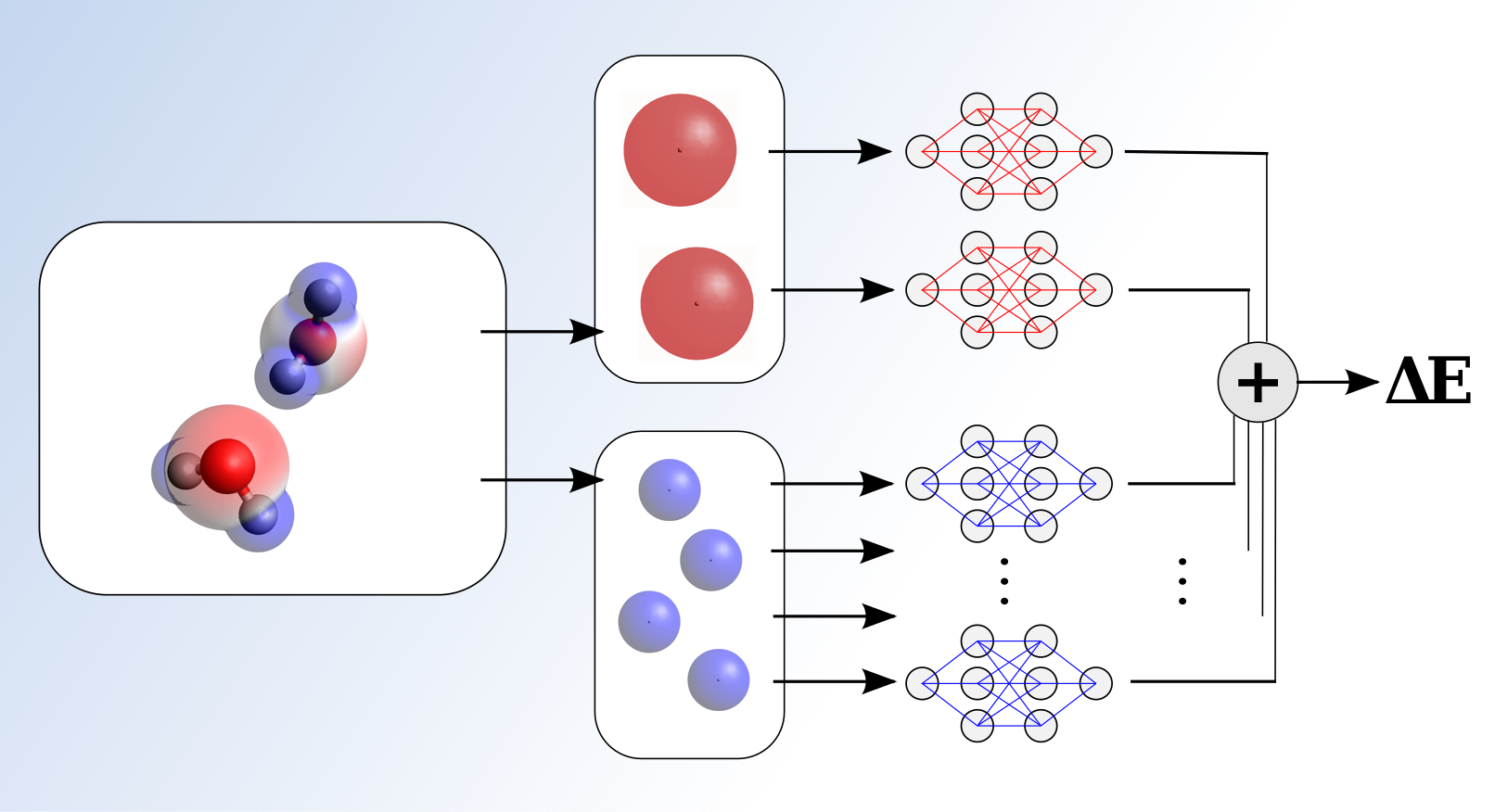
We propose a new molecular simulation framework that combines the transferability, robustness and chemical flexibility of an ab initio method with the accuracy and efficiency of a machine learned force field. The key to achieve this mix is to use a standard density functional theory (DFT) simulation as a pre-processor for the atomic and molecular information, obtaining a good quality electronic density. General, symmetry preserving, atom-centered electronic descriptors are then built from this density to train a neural network to correct the baseline DFT energies and forces. These electronic descriptors encode much more information than local atomic environments, allowing a simple neural network to reach the accuracy required for the problem of study at a negligible cost. The balance between accuracy and efficiency is determined by the baseline simulation. This is shown in results where high level quantum chemical accuracy is obtained for simulations of liquid water at standard DFT cost, or where high level DFT-accuracy is achieved in simulations with a low-level baseline DFT calculation, at a significantly reduced cost.
PDF Abstract