Molecular Mechanics-Driven Graph Neural Network with Multiplex Graph for Molecular Structures
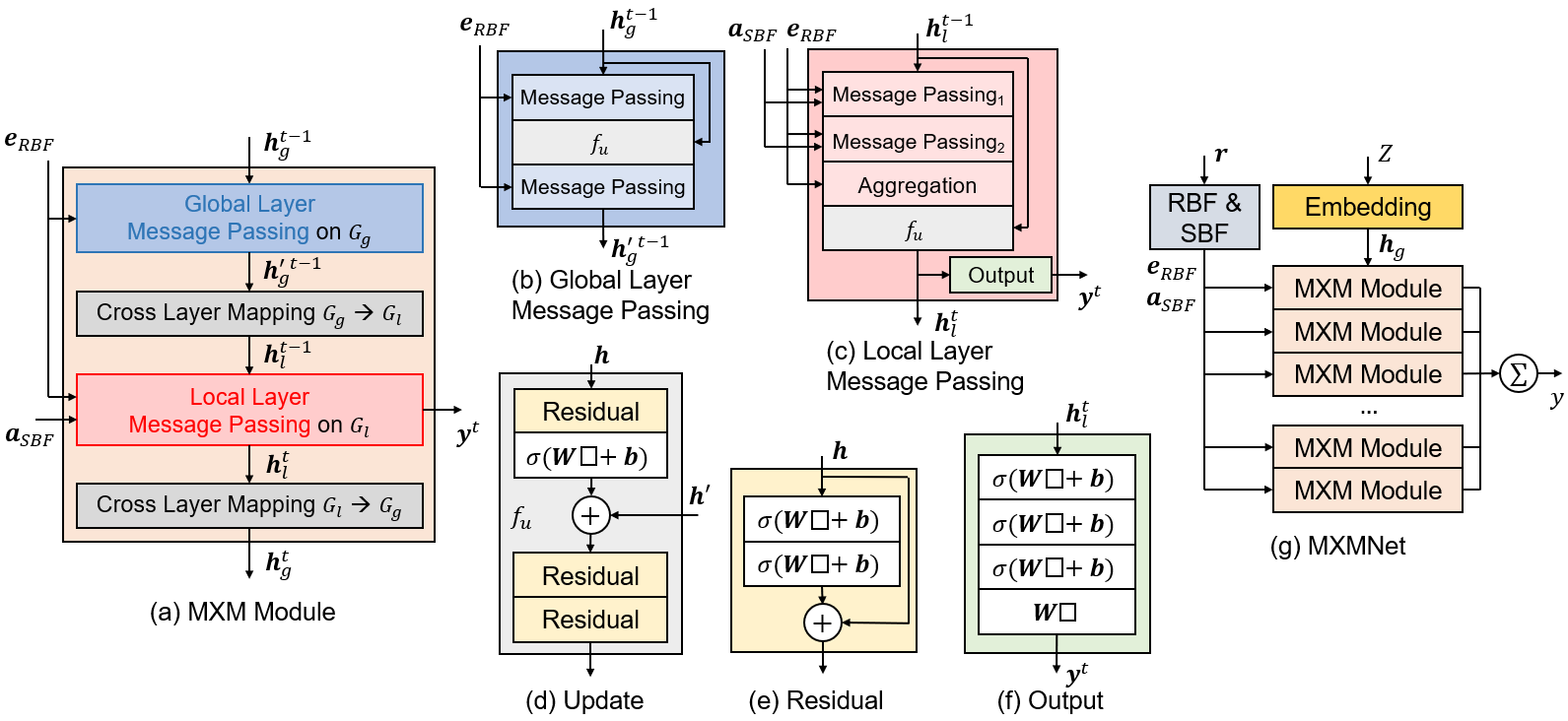
The prediction of physicochemical properties from molecular structures is a crucial task for artificial intelligence aided molecular design. A growing number of Graph Neural Networks (GNNs) have been proposed to address this challenge. These models improve their expressive power by incorporating auxiliary information in molecules while inevitably increase their computational complexity. In this work, we aim to design a GNN which is both powerful and efficient for molecule structures. To achieve such goal, we propose a molecular mechanics-driven approach by first representing each molecule as a two-layer multiplex graph, where one layer contains only local connections that mainly capture the covalent interactions and another layer contains global connections that can simulate non-covalent interactions. Then for each layer, a corresponding message passing module is proposed to balance the trade-off of expression power and computational complexity. Based on these two modules, we build Multiplex Molecular Graph Neural Network (MXMNet). When validated by the QM9 dataset for small molecules and PDBBind dataset for large protein-ligand complexes, MXMNet achieves superior results to the existing state-of-the-art models under restricted resources.
PDF Abstract


 QM9
QM9

