Advanced Graph and Sequence Neural Networks for Molecular Property Prediction and Drug Discovery
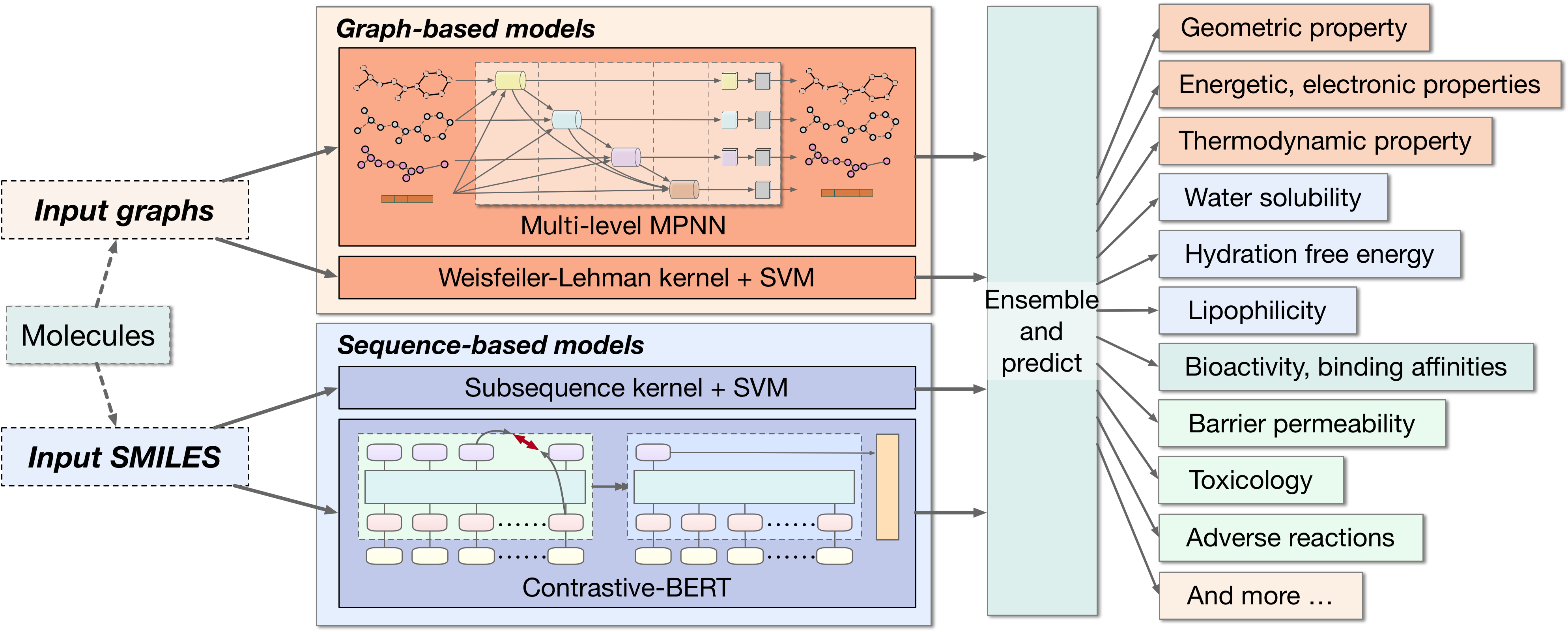
Properties of molecules are indicative of their functions and thus are useful in many applications. With the advances of deep learning methods, computational approaches for predicting molecular properties are gaining increasing momentum. However, there lacks customized and advanced methods and comprehensive tools for this task currently. Here we develop a suite of comprehensive machine learning methods and tools spanning different computational models, molecular representations, and loss functions for molecular property prediction and drug discovery. Specifically, we represent molecules as both graphs and sequences. Built on these representations, we develop novel deep models for learning from molecular graphs and sequences. In order to learn effectively from highly imbalanced datasets, we develop advanced loss functions that optimize areas under precision-recall curves. Altogether, our work not only serves as a comprehensive tool, but also contributes towards developing novel and advanced graph and sequence learning methodologies. Results on both online and offline antibiotics discovery and molecular property prediction tasks show that our methods achieve consistent improvements over prior methods. In particular, our methods achieve #1 ranking in terms of both ROC-AUC and PRC-AUC on the AI Cures Open Challenge for drug discovery related to COVID-19. Our software is released as part of the MoleculeX library under AdvProp.
PDF Abstract


