Pocket2Mol: Efficient Molecular Sampling Based on 3D Protein Pockets
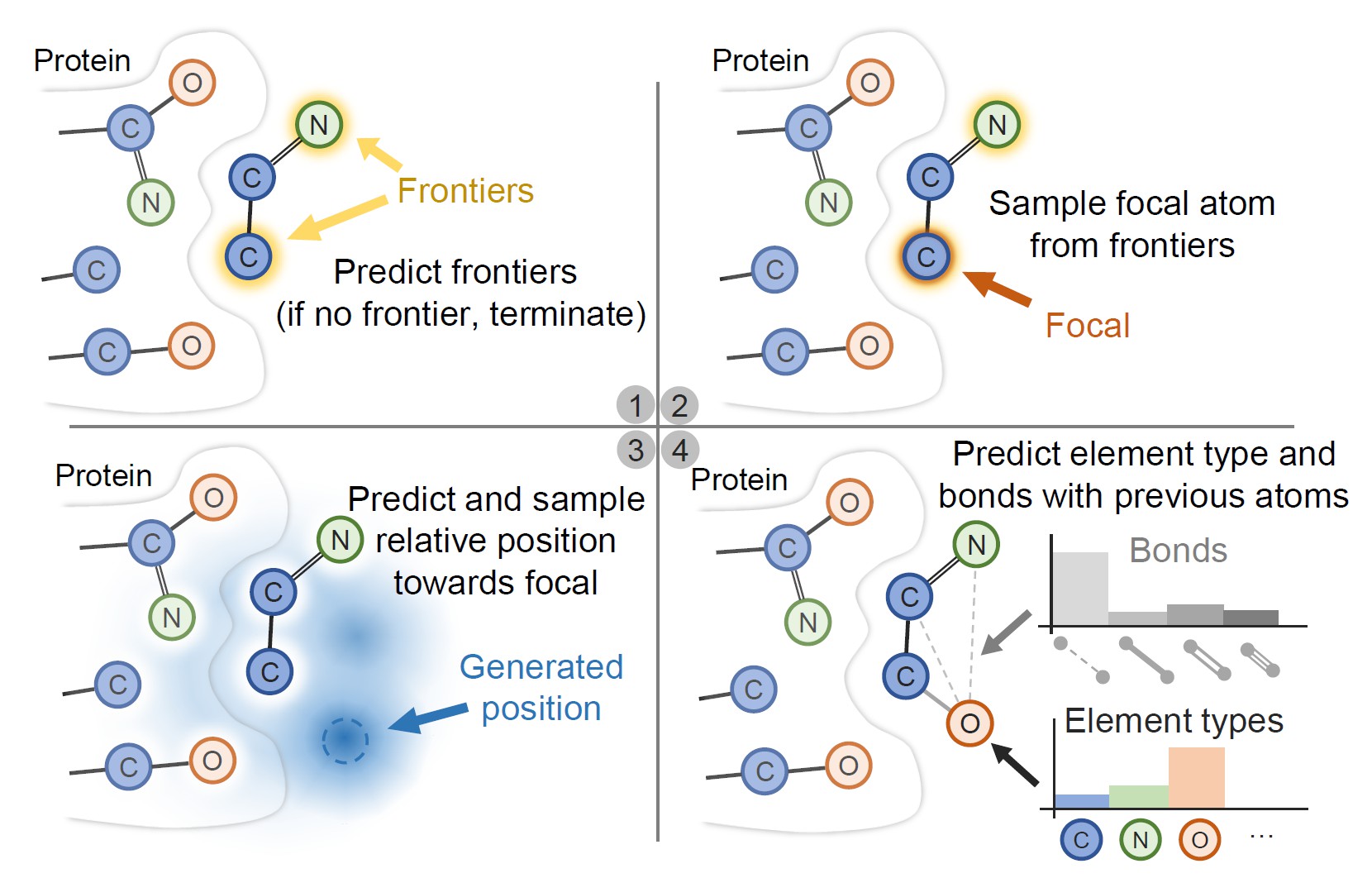
Deep generative models have achieved tremendous success in designing novel drug molecules in recent years. A new thread of works have shown the great potential in advancing the specificity and success rate of in silico drug design by considering the structure of protein pockets. This setting posts fundamental computational challenges in sampling new chemical compounds that could satisfy multiple geometrical constraints imposed by pockets. Previous sampling algorithms either sample in the graph space or only consider the 3D coordinates of atoms while ignoring other detailed chemical structures such as bond types and functional groups. To address the challenge, we develop Pocket2Mol, an E(3)-equivariant generative network composed of two modules: 1) a new graph neural network capturing both spatial and bonding relationships between atoms of the binding pockets and 2) a new efficient algorithm which samples new drug candidates conditioned on the pocket representations from a tractable distribution without relying on MCMC. Experimental results demonstrate that molecules sampled from Pocket2Mol achieve significantly better binding affinity and other drug properties such as druglikeness and synthetic accessibility.
PDF Abstract

