Molecular Docking
21 papers with code • 0 benchmarks • 0 datasets
Predicting the binding structure of a small molecule ligand to a protein, which is critical to drug design.
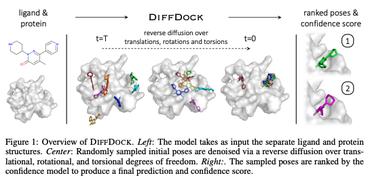
Description from: DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking
Benchmarks
These leaderboards are used to track progress in Molecular Docking
Libraries
Use these libraries to find Molecular Docking models and implementations
Most implemented papers
SVSBI: Sequence-based virtual screening of biomolecular interactions
weilabmsu/svs
•
 •
•
Virtual screening (VS) is an essential technique for understanding biomolecular interactions, particularly, drug design and discovery.
Domain-Agnostic Molecular Generation with Chemical Feedback
zjunlp/MolGen
•
•
The generation of molecules with desired properties has become increasingly popular, revolutionizing the way scientists design molecular structures and providing valuable support for chemical and drug design.
Implicit Geometry and Interaction Embeddings Improve Few-Shot Molecular Property Prediction
cfifty/ignite
•
•
However, many important molecular properties depend on complex molecular characteristics -- such as the various 3D geometries a molecule may adopt or the types of chemical interactions it can form -- that are not explicitly encoded in the feature space and must be approximated from low amounts of data.
Modeling Molecular Structures with Intrinsic Diffusion Models
gcorso/diffdock
•
•
Since its foundations, more than one hundred years ago, the field of structural biology has strived to understand and analyze the properties of molecules and their interactions by studying the structure that they take in 3D space.
Optimization of binding affinities in chemical space with generative pretrained transformer and deep reinforcement learning
charlesxu90/sgpt
•
•
F1000Research 2023
The SGPT-RL method achieved better results than Reinvent on the ACE2 task, where molecular docking was used as the optimization goal.
Transferable Graph Neural Fingerprint Models for Quick Response to Future Bio-Threats
With this dataset, we trained graph neural fingerprint docking models for high-throughput virtual COVID-19 drug screening.
PLANTAIN: Diffusion-inspired Pose Score Minimization for Fast and Accurate Molecular Docking
molecularmodelinglab/plantain
•
•
Traditional docking methods predict ligand poses by minimizing a physics-inspired scoring function.
DiffBindFR: An SE(3) Equivariant Network for Flexible Protein-Ligand Docking
Furthermore, in the Apo and AlphaFold2 modeled structures, DiffBindFR demonstrates superior advantages in accurate ligand binding pose and protein binding conformation prediction, making it suitable for Apo and AlphaFold2 structure-based drug design.
Enhancing Ligand Pose Sampling for Molecular Docking
To train scoring functions-and to perform molecular docking-one must generate a set of candidate ligand binding poses.
Equivariant Scalar Fields for Molecular Docking with Fast Fourier Transforms
The runtime of our approach can be amortized at several levels of abstraction, and is particularly favorable for virtual screening settings with a common binding pocket.
