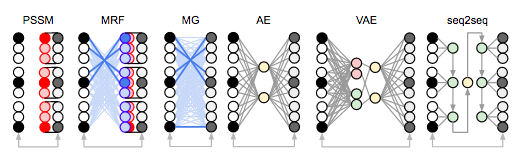
Unified framework for modeling multivariate distributions in biological sequences
Revealing the functional sites of biological sequences, such as evolutionary conserved, structurally interacting or co-evolving protein sites, is a fundamental, and yet challenging task. Different frameworks and models were developed to approach this challenge, including Position-Specific Scoring Matrices, Markov Random Fields, Multivariate Gaussian models and most recently Autoencoders. Each of these methods has certain advantages, and while they have generated a set of insights for better biological predictions, these have been restricted to the corresponding methods and were difficult to translate to the complementary domains. Here we propose a unified framework for the above-mentioned models, that allows for interpretable transformations between the different methods and naturally incorporates the advantages and insight gained individually in the different communities. We show how, by using the unified framework, we are able to achieve state-of-the-art performance for protein structure prediction, while enhancing interpretability of the prediction process.
PDF Abstract
