Autoregressive neural-network wavefunctions for ab initio quantum chemistry
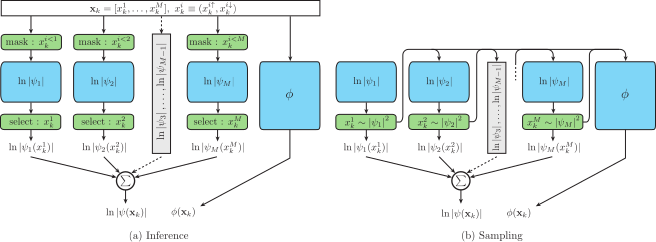
In recent years, neural network quantum states (NNQS) have emerged as powerful tools for the study of quantum many-body systems. Electronic structure calculations are one such canonical many-body problem that have attracted significant research efforts spanning multiple decades, whilst only recently being attempted with NNQS. However, the complex non-local interactions and high sample complexity are significant challenges that call for bespoke solutions. Here, we parameterise the electronic wavefunction with a novel autoregressive neural network (ARN) that permits highly efficient and scalable sampling, whilst also embedding physical priors reflecting the structure of molecular systems without sacrificing expressibility. This allows us to perform electronic structure calculations on molecules with up to 30 spin-orbitals -- at least an order of magnitude more Slater determinants than previous applications of conventional NNQS -- and we find that our ansatz can outperform the de-facto gold-standard coupled cluster methods even in the presence of strong quantum correlations. With a highly expressive neural network for which sampling is no longer a computational bottleneck, we conclude that the barriers to further scaling are not associated with the wavefunction ansatz itself, but rather are inherent to any variational Monte Carlo approach.
PDF Abstract Colab
Colab


