DockGame: Cooperative Games for Multimeric Rigid Protein Docking
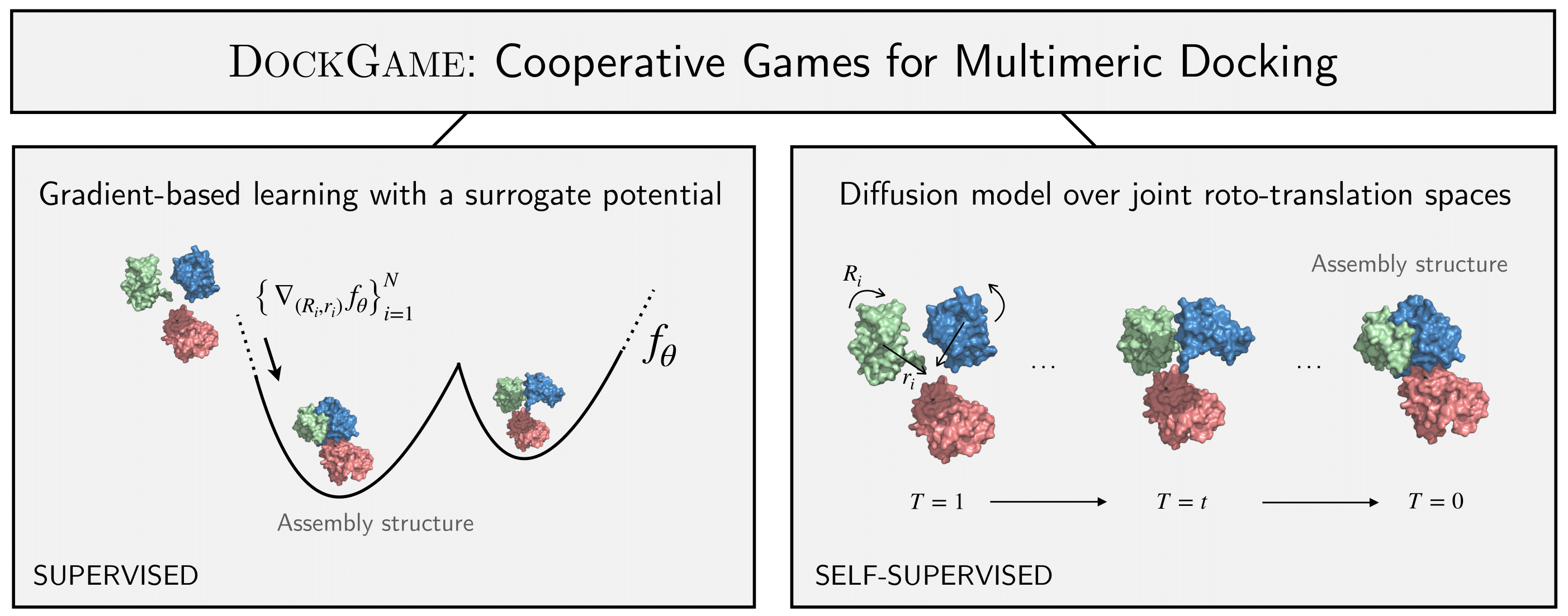
Protein interactions and assembly formation are fundamental to most biological processes. Predicting the assembly structure from constituent proteins -- referred to as the protein docking task -- is thus a crucial step in protein design applications. Most traditional and deep learning methods for docking have focused mainly on binary docking, following either a search-based, regression-based, or generative modeling paradigm. In this paper, we focus on the less-studied multimeric (i.e., two or more proteins) docking problem. We introduce DockGame, a novel game-theoretic framework for docking -- we view protein docking as a cooperative game between proteins, where the final assembly structure(s) constitute stable equilibria w.r.t. the underlying game potential. Since we do not have access to the true potential, we consider two approaches - i) learning a surrogate game potential guided by physics-based energy functions and computing equilibria by simultaneous gradient updates, and ii) sampling from the Gibbs distribution of the true potential by learning a diffusion generative model over the action spaces (rotations and translations) of all proteins. Empirically, on the Docking Benchmark 5.5 (DB5.5) dataset, DockGame has much faster runtimes than traditional docking methods, can generate multiple plausible assembly structures, and achieves comparable performance to existing binary docking baselines, despite solving the harder task of coordinating multiple protein chains.
PDF Abstract

