Machine-learned molecular mechanics force field for the simulation of protein-ligand systems and beyond
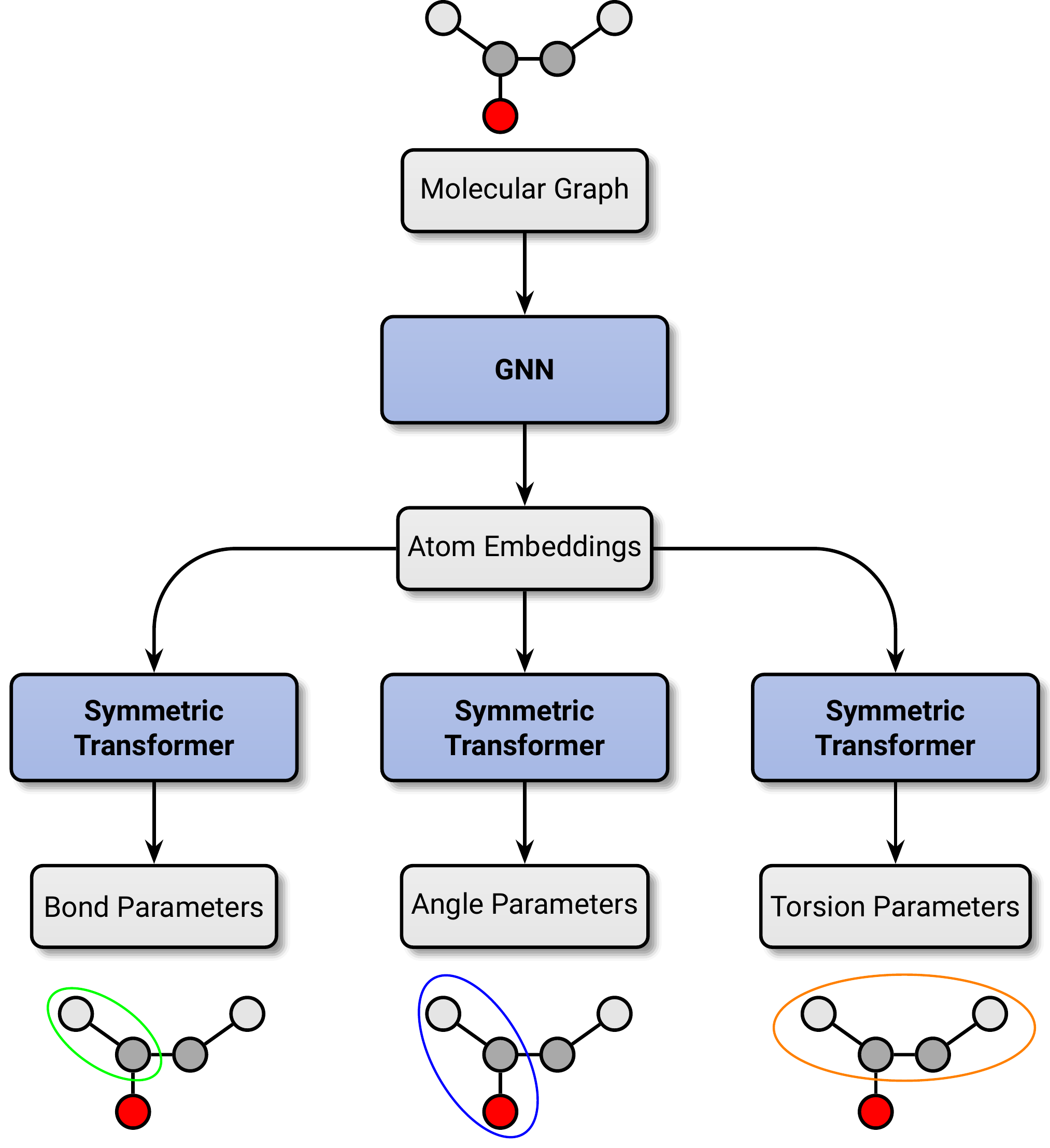
The development of reliable and extensible molecular mechanics (MM) force fields -- fast, empirical models characterizing the potential energy surface of molecular systems -- is indispensable for biomolecular simulation and computer-aided drug design. Here, we introduce a generalized and extensible machine-learned MM force field, \texttt{espaloma-0.3}, and an end-to-end differentiable framework using graph neural networks to overcome the limitations of traditional rule-based methods. Trained in a single GPU-day to fit a large and diverse quantum chemical dataset of over 1.1M energy and force calculations, \texttt{espaloma-0.3} reproduces quantum chemical energetic properties of chemical domains highly relevant to drug discovery, including small molecules, peptides, and nucleic acids. Moreover, this force field maintains the quantum chemical energy-minimized geometries of small molecules and preserves the condensed phase properties of peptides, self-consistently parametrizing proteins and ligands to produce stable simulations leading to highly accurate predictions of binding free energies. This methodology demonstrates significant promise as a path forward for systematically building more accurate force fields that are easily extensible to new chemical domains of interest.
PDF Abstract

