Graph Denoising Diffusion for Inverse Protein Folding
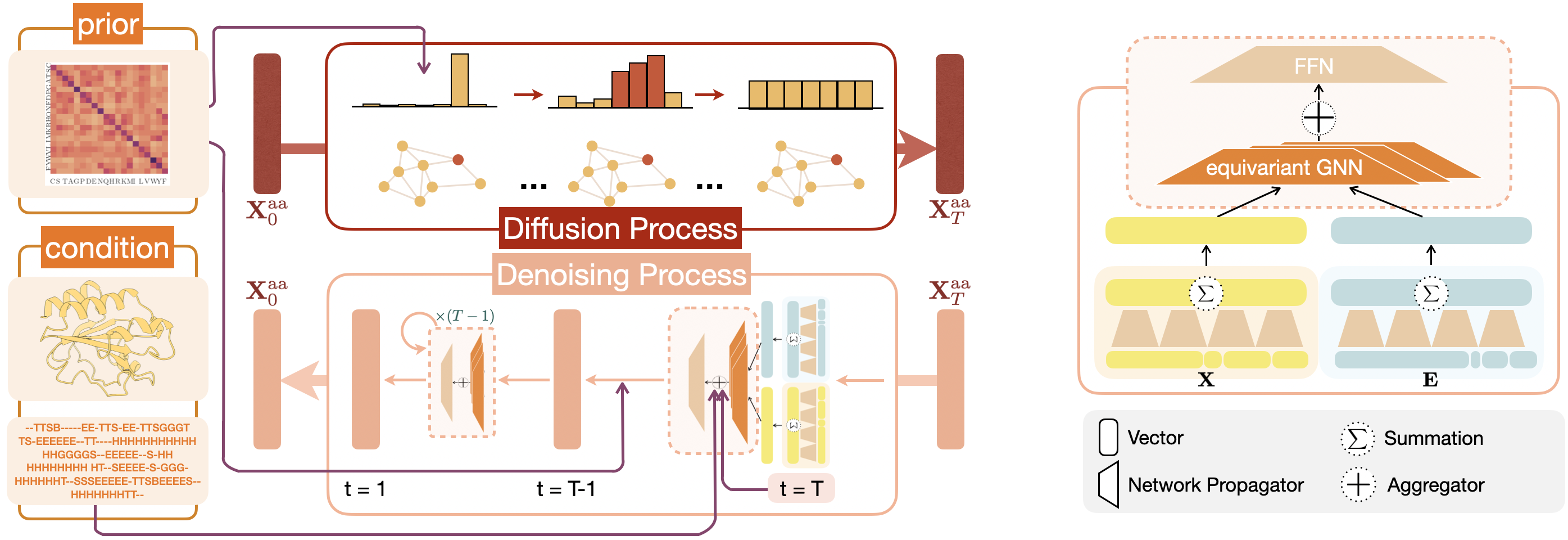
Inverse protein folding is challenging due to its inherent one-to-many mapping characteristic, where numerous possible amino acid sequences can fold into a single, identical protein backbone. This task involves not only identifying viable sequences but also representing the sheer diversity of potential solutions. However, existing discriminative models, such as transformer-based auto-regressive models, struggle to encapsulate the diverse range of plausible solutions. In contrast, diffusion probabilistic models, as an emerging genre of generative approaches, offer the potential to generate a diverse set of sequence candidates for determined protein backbones. We propose a novel graph denoising diffusion model for inverse protein folding, where a given protein backbone guides the diffusion process on the corresponding amino acid residue types. The model infers the joint distribution of amino acids conditioned on the nodes' physiochemical properties and local environment. Moreover, we utilize amino acid replacement matrices for the diffusion forward process, encoding the biologically-meaningful prior knowledge of amino acids from their spatial and sequential neighbors as well as themselves, which reduces the sampling space of the generative process. Our model achieves state-of-the-art performance over a set of popular baseline methods in sequence recovery and exhibits great potential in generating diverse protein sequences for a determined protein backbone structure.
PDF Abstract NeurIPS 2023 PDF NeurIPS 2023 Abstract


