Applying Deep Reinforcement Learning to the HP Model for Protein Structure Prediction
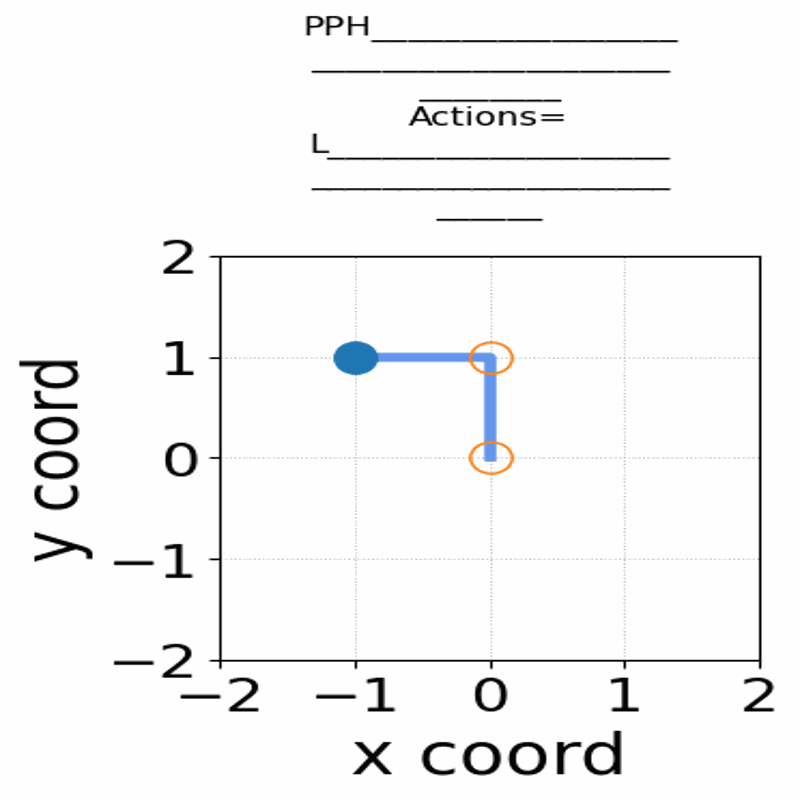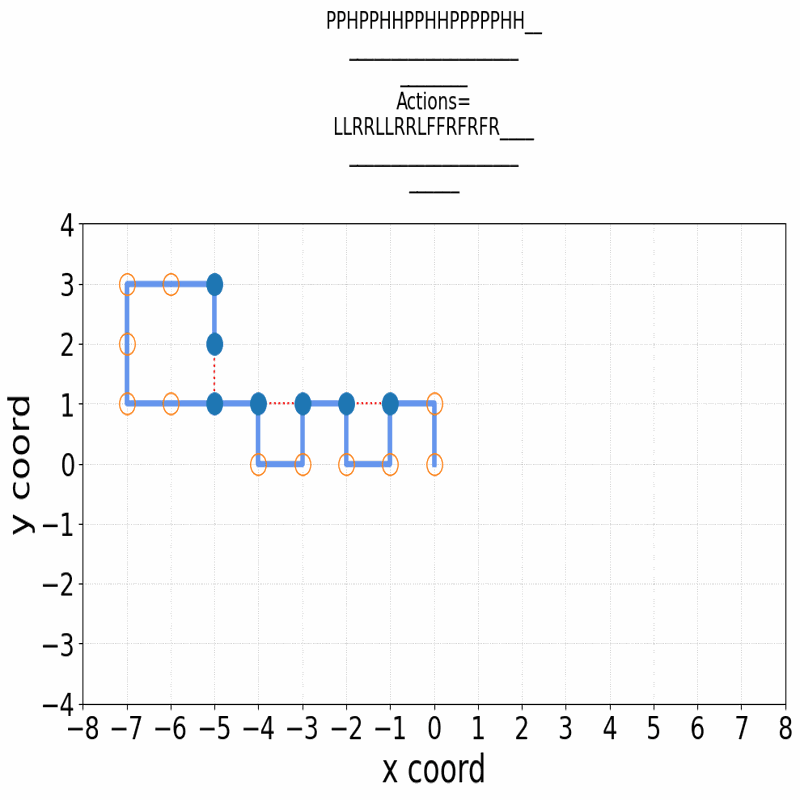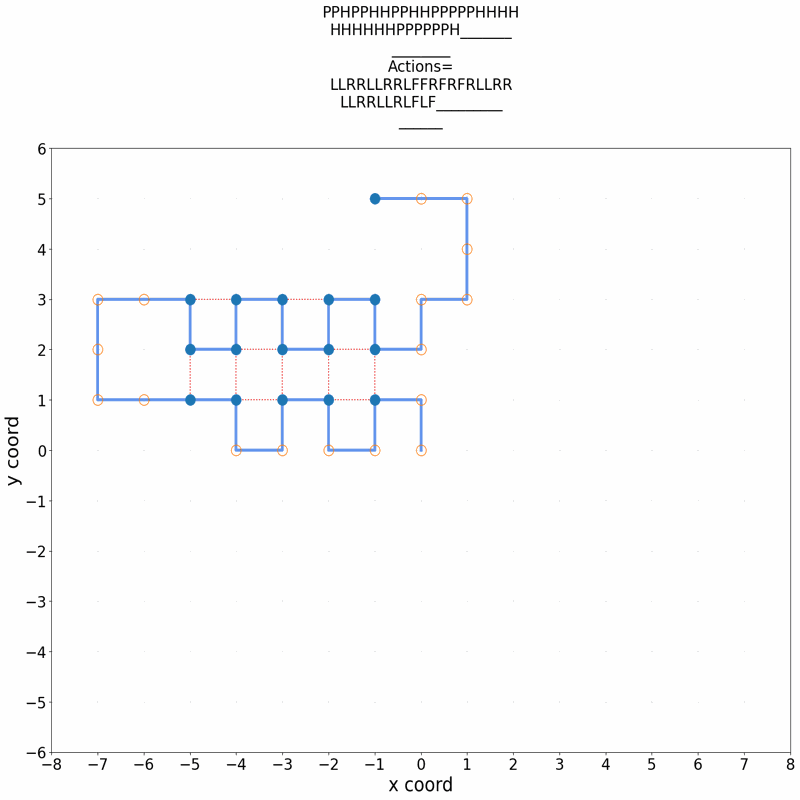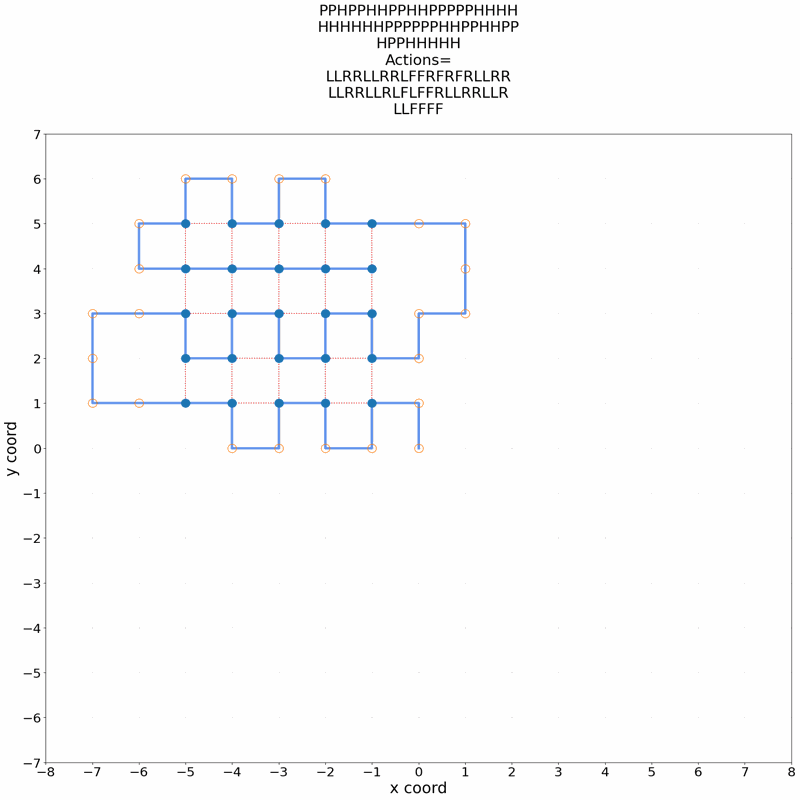
A central problem in computational biophysics is protein structure prediction, i.e., finding the optimal folding of a given amino acid sequence. This problem has been studied in a classical abstract model, the HP model, where the protein is modeled as a sequence of H (hydrophobic) and P (polar) amino acids on a lattice. The objective is to find conformations maximizing H-H contacts. It is known that even in this reduced setting, the problem is intractable (NP-hard). In this work, we apply deep reinforcement learning (DRL) to the two-dimensional HP model. We can obtain the conformations of best known energies for benchmark HP sequences with lengths from 20 to 50. Our DRL is based on a deep Q-network (DQN). We find that a DQN based on long short-term memory (LSTM) architecture greatly enhances the RL learning ability and significantly improves the search process. DRL can sample the state space efficiently, without the need of manual heuristics. Experimentally we show that it can find multiple distinct best-known solutions per trial. This study demonstrates the effectiveness of deep reinforcement learning in the HP model for protein folding.
PDF Abstract